Câncer de Adrenal
Os tumores de adrenal são divididos em tumores da cortical (ou carcinoma adrenocortical) e tumores da medular (feocromocitomas).
Câncer de adrenal (carcinoma adrenocortical)
Introdução
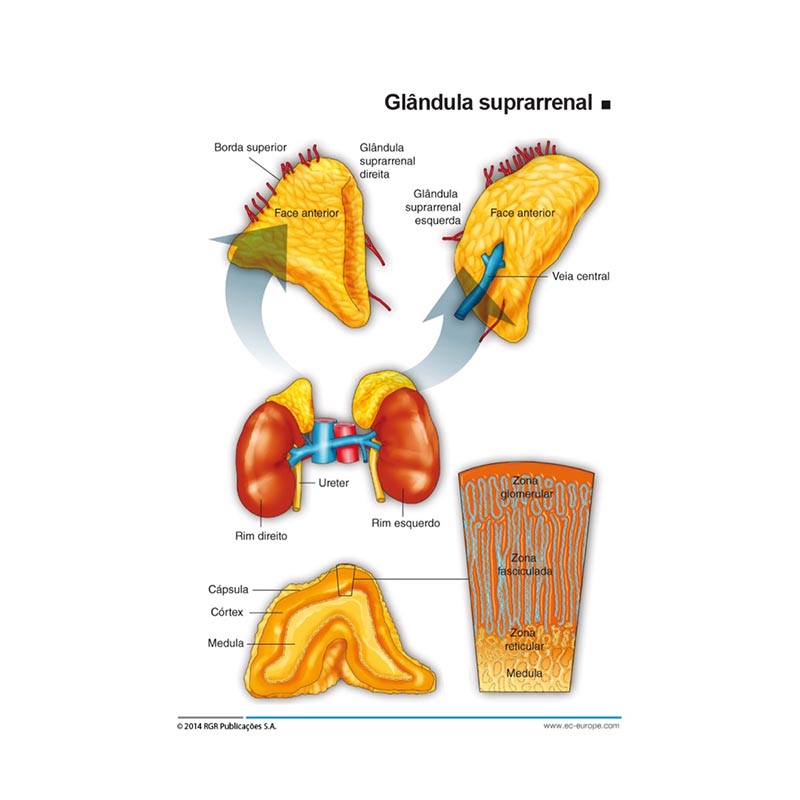
O câncer de adrenal é originado na córtex da glândula adrenal (ou suprerrenal). É um tumor infreqüente, com incidência anual de 1-2 casos/1.000.000 (0,02% de todas neoplasias, 0,2% dos óbitos por câncer). Muito menos freqüente que os adenomas corticais benignos. Incidência similar entre os sexos, com três picos de ocorrência reconhecidos (em crianças de 1 e 2 anos, entre 7 e 16 anos, e mais comumente em adultos na 5ª década de vida). São tumores redominantes da adrenal esquerda (50-70%), sendo bilaterais em 5-7% dos casos no momento do diagnóstico. Podem ser descritos como parte de síndromes hereditárias complexas que incluem sarcomas, câncer de mama e pulmão. São tumores bastante agressivos com tendência a invadir estruturas adjacentes.
Clínica
Os sintomas mais comuns do câncer de adrenal são: dor abdominal (50%), fadiga (25%), massa abdominal (25%), perda de peso (15%), hematúria (sangramento da urina) (7%), febre (7%) e sintomas referentes ao excesso de esteróides, tais como acnes, oligomenorréia e fragilidade cutênea.
O câncer de adrenal pode ser classificado, do ponto de vista endocrinológico, como funcionante ou não-funcionante (na proporção de 1:1 em homens e 2:1 em mulheres). Os tumores funcionantes geralmente manifestam-se como síndromes clínicas por excesso de produção de hormônios (Síndrome de Cushing [hipercortisolismo] em 67%, Síndrome de Conn [hiperalsoteronismo] em 7%, Virilização/Feminização/Puberdade precoce em 11%, ou uma combinação destas em 15%). Assim, uma meticulosa avaliação endocrinológica se faz necessária.
O diagnóstico do câncer de adrenal ocorre geralmente quando a doença alcança volumes tumorais maiores que 6-20cm, devido à sua localização retroperitoneal. A ressonância nuclear magnética (RNM) é o método mais preciso para avaliar a extensão local e os linfonodos (gânglios).
As vias de disseminação mais freqüentes do câncer de adrenal são linfática ou hematogênica. Os locais mais freqüentes para metástases são os gânglios linfáticos (68%), pulmão (71%), fígado (42%) e óssos (26%).
O estadiamento (estudo da extensão da doença) é realizado com ultrassonografia de abdome (hepático), radiografia de tórax e, excepcionalmente, tomografia computadorizada de tórax (em casos de alterações na radiografia) e cintilografia óssea (em casos de dor óssea ou elevação do marcador tumoral fosfatase alcalina). RNM, ultrassonografia com doppler e ultrassonografia transesofágica podem ser úteis para avaliar possíveis trombos na veia cava.
| Estadiamento TNM (AJCC, 7th ed., 2010) | ||
|---|---|---|
| T | Tx | O tumor primário não pode ser avaliado |
| T0 | Não há evidência de tumor primário | |
| T1 | Tumor 5 centímetros ou menos em sua maior dimensão, sem invasão extra-adrenal | |
| T2 | Tumor maior que 5 cm, invasão não extra-adrenal | |
| T3 | Tumor de qualquer tamanho, com invasão local, mas não invadir órgãos adjacentes | |
| T4 | Tumor de qualquer tamanho com invasão de órgãos adjacentes* | |
| N | NX | Os linfonodos não podem ser avaliados |
| N0 | Sem metástases em linfonodos regionais | |
| N1 | Metástases em linfonodo(s) regional(is) | |
| M | M0 | Sem metástases à distância |
| M1 | Metástases à distância | |
*Órgãos adjacentes incluem rim, diafragma, grandes vasos, pâncreas, baço e fígado
pTNM Classificação Patológica
As categorias pT, pN e pM correspondem para as categorias T, N e M, com excepção de que pM0 não existe como uma categoria.
pN0
O exame histológico de uma linfadenectomia regional incluirá, geralmente, 12 ou mais linfonodos. Se os nódulos linfáticos são negativos, mas o número normalmente examinado não for atendido, classificar como pN0.
| Grupamento por Estádios | |||
| Estádio | TNM | Sullivan (modificado de Mac Farlane) | Nader |
| I | T1 N0 M0 | Tumor < 5 cm confinado à glândula adrenal | Tumor confinado à glândula adrenal sem evidência de extensão local ou à distância |
| II | T2 N0 M0 | Tumor > 5 cm confinado à glândula adrenal | Tumor afeta os gânglios linfáticos regionais e tecidos adjacentes ou órgãos vizinhos |
| III | T1-2 N1 M0T3 N0 M0 | Invasão local ou gânglios positivos | Metástases à distância viscerais, ósseas ou em SNC |
| IV | T4 N0 M0T3-4 N1 M0T1-4 N0-1 M1 | Invasão local com gânglios positivos ou metástases à distância | |
Tratamento
O tratamento do câncer de adrenal é eminentemente cirúrgico, com ressecções agressivas e radicais em todos os estádios, quer seja por via aberta ou videolaparoscópica. A ressecção cirúrgica radical indica-se em todos incidentalomas (tumor identificado em exames de rotina, assintomático) maiores que 4-5cm, tumores em estádio I e II. Medidas de suporte (quimioterapia e/ou radioterapia) estão indicadas nos tumores estádio III e IV e nos tumores irressecáveis ou com contra-indicação cirúrgica.
Seu prognóstico é limitado, com índice de sobrevida média de 2 a 3 anos, sendo a sobrevida global em 5 anos de 25-30% (Estádio I: 45-50%, II: 10-15% e III-IV: 5%).
Seguimento
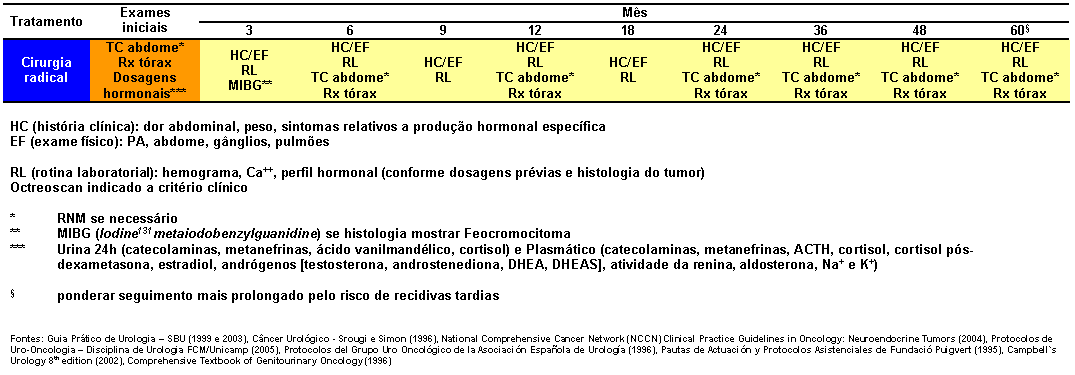
Feocromocitomas / Paragangliomas
Introdução
Feocromocitomas são neoplasias da medula da glândula adrenal (suprarrenal), nela situados em 80-90% dos casos. Paragangliomas são feocromocitomas ectópicos ou extra-adrenais que surgem a partir de gânglios simpáticos para-aórticos. Feocromocitomas e paragangliomas ocorrer em 0,05% a 0,1% dos pacientes hipertensos, e são extremamente raros.
Clínica
Feocromocitomas liberam catecolaminas e metabólitos da adrenalina e noradrenalina, resultando em hipertensão, arritmia e/ou hiperglicemia. Cerca de 40% dos paragangliomas também secretam catecolaminas. O pico de incidência dos feocromocitomas é entre a terceira e a quinta décadas de vida, mas geralmente ocorrem em uma idade mais jovem, e são mais propensos à bilateralidade em pacientes com doença familiar, sendo malignos em 10% dos casos. Já os paragangliomas são malignos em aproximadamente 40% dos casos. Quando associados a síndromes familiares, tendem a ser mais precoces, agressivos e com maior probabilidade de metástase. Assim, pacientes que apresentam esses tumores durante a infância, adolescência ou idade adulta jovem requerem cuidado e vigilância ao longo da vida.
Pacientes com suspeita de feocromocitoma devem ser avaliados com dosagens laboratoriais (no sangue e urina) de possível metabólitos da adrenalina e noradrenalina, e catecolaminas, embora 15-20% dos doentes com feocromocitoma apresentem níveis normais de catecolaminas da urina, devido a secreção intermitente em alguns tumores e secreção insignificante pelos outros. Níveis muito elevados são, em geral, diagnósticos. A medição dos níveis de dopamina deve ser indicada para paragangliomas cervicais. Exames de imagem recomendados incluem tomografia computadorizada de tórax e ressonância nuclear magnética de abdome. A cintilografia com metaiodobenzilguanidina (MIBG) é bastante útil para localizar feocromocitomas (incluindo tumores extra-adrenais), principalmente em tumores de difícil identificação. Já a cintilografia com somatostatina é opcional (suspeita de tumores múltiplos). A cintilografia óssea deve ser realizada em casos de suspeita clínica.
Apesar de 59-90% dos casos de feocromocitoma serem doenças esporádicas, podem ocorrem com frequência em pacientes com doenças ou síndromes familiares. Pacientes com idade Quanto ao tratamento dos feocromocitomas e paragangliomas, a ressecção cirúrgica é a terapia fundamental, tanto nos tumores malignos quanto benignos. Entretanto, antes da cirurgia os pacientes devem ser medicados com bloqueadores alfa-adrenérgicos (como fenoxibenzamida ou doxazosina) para reduzir o risco de crises hipertensivas, bem como bloqueadores beta-adrenérgicos (como propranolol, atenolol ou metoprolol).
| Estadiamento TNM (AJCC, 7th ed., 2010) | ||
|---|---|---|
| T | Tx | O tumor primário não pode ser avaliado |
| T0 | Não há evidência de tumor primário | |
| T1 | Tumor 5 centímetros ou menos em sua maior dimensão, sem invasão extra-adrenal | |
| T2 | Tumor maior que 5 cm, invasão não extra-adrenal | |
| T3 | Tumor de qualquer tamanho, com invasão local, mas não invadir órgãos adjacentes | |
| T4 | Tumor de qualquer tamanho com invasão de órgãos adjacentes* | |
| N | NX | Os linfonodos não podem ser avaliados |
| N0 | Sem metástases em linfonodos regionais | |
| N1 | Metástases em linfonodo(s) regional(is) | |
| M | M0 | Sem metástases à distância |
| M1 | Metástases à distância | |
*Órgãos adjacentes incluem rim, diafragma, grandes vasos, pâncreas, baço e fígado
pTNM Classificação Patológica
As categorias pT, pN e pM correspondem para as categorias T, N e M, com excepção de que pM0 não existe como uma categoria.
pN0
O exame histológico de uma linfadenectomia regional incluirá, geralmente, 12 ou mais linfonodos. Se os nódulos linfáticos são negativos, mas o número normalmente examinado não for atendido, classificar como pN0.
| Grupamento por Estádios | |||
| Estádio | TNM | Sullivan (modificado de Mac Farlane) | Nader |
| I | T1 N0 M0 | Tumor < 5 cm confinado à glândula adrenal | Tumor confinado à glândula adrenal sem evidência de extensão local ou à distância |
| II | T2 N0 M0 | Tumor > 5 cm confinado à glândula adrenal | Tumor afeta os gânglios linfáticos regionais e tecidos adjacentes ou órgãos vizinhos |
| III | T1-2 N1 M0T3 N0 M0 | Invasão local ou gânglios positivos | Metástases à distância viscerais, ósseas ou em SNC |
| IV | T4 N0 M0T3-4 N1 M0T1-4 N0-1 M1 | Invasão local com gânglios positivos ou metástases à distância | |
Tratamento
A abordagem videolaparoscópica, quando segura e viável, é o tratamento de escolha para os feocromocitomas adrenais.
Em casos de doença localmente avançada ou metastática, tanto as lesões primárias quanto as metástases isoladas devem ser tratadas cirurgicamente, quanto possível, numa abordagem citorredutora, com ou sem radioterapia. Quando a cirurgia não for factível, opções terapêuticas incluem quimioterapia sistêmica ou terapia com iodo-131-MIBG (em tumores que captam MIBG).
O seguimento dos pacientes com feocromocitoma ou paragangliomas são semelhantes aos de outros tumores neuroendócrinos. Após a ressecção completa, história clínica e exame físico (com medida da pressão arterial) devem ser realizadas a cada seis meses nos primeiros 3 anos, e anualmente por até 10 anos. Os pacientes com doença persistente necessitam exame mais frequentes, em intervalos de cada 3 a 4 meses. Além disso, exames de tomografia computadorizada, ressonância nuclear magnética, ou FDG-PET podem ser considerado. Testes e aconselhamento genético também são recomendados quando clinicamente indicados.
Seguimento
Fontes:
Campbell-Walsh Urology, 10th edition (2011)
Comprehensive Textbook of Genitourinary Oncology, 4th edition (2011)
National Comprehensive Cancer Network (NCCN) Clinical Practice Guidelines in Oncology (NCCN Guidelines®) – Neuroendocrine Tumors – Version 2.2014
Warning: Attempt to read property "term_id" on bool in /home/u115818844/domains/neouro.com.br/public_html/wp-content/themes/neouro/single-tratamentos.php on line 31