Câncer de Retroperitônio
Introdução
Os tumores de retroperitônio representam 0,2% de todas as neoplasias diagnosticadas atualmente. São tumores típicos da 4° a 6° décadas (podendo ocorrer em qualquer idade), com ligeiro predomínio masculino. Têm origem mesenquimal (vascular, muscular, tecido conectivo, tecido adiposo, tecido neural e remanecentes embriônicos), sendo que 1/3 dos tumores malignos originários do retroperitônio são sarcomas, e aproximadamente 15% dos sarcomas de partes moles originam-se no retroperitônio.
Estão associados a fatores de risco como radioterapia prévia (principalmente MFH [histiocitoma fibroso maligno], osteosarcoma e fibrosarcoma), trauma, exposição ao asbesto e herbicidas.
Os tumores de retroperitônio são malignos em 70-80% dos casos, sendo 50% de alto grau. Dentre as lesões benignas destacam-se o lipoma (mais comum), lipomatose pélvica, mielolipoma (geralmente na glândula adrenal), xantogranuloma (hoje subclassificado como MFH), leiomioma, ganglioneuroma, homangiopericitoma e schwanoma. Das lesões malignas temos o lipossarcoma (mais comum, podendo metastatizar) em 41% dos casos, leiomiossarcoma em 28%, MFH (cada vez mais comum por diagnóstico molecular) em 7% dos casos, fibrosarcoma (alto risco de metástases) em 6%, tumores malignos da bainha neural periférica em 3%, além de rabdomiosarcoma (significante em pediatria), hemangiopericitoma maligno, sarcoma de células sinoviais e angiossarcoma.
Clínica
Quando os tumores de retroperitônio são menores de 5cm, raramente são percebidos pelos pacientes, até que atinjam tamanhos maiores. Como manifestações clínicas, apresentam principalmente massa abdominal e perda de peso, dor abdominal vaga e difusa. O diagnóstico diferencial é com hematoma (geralmente associado à história de trauma ou anticoagulantes), abscessos (que cursam com febre, leucocitose) e metástase ou cistos (rim, pâncreas, adrenal). Deve-se descartar a presença de tumores funcionantes e não-funcionantes de adrenal (com provas funcionais), tumores renais, pancreáticos, gastrointestinais (com endoscopia digestiva), de células germinativas (com exame testicular e marcadores tumorais) e linfomas (com biópsia excisional). Uma história clínica e exame físico detalhados podem ajudar a distinguir tais enfermidades. Em lesões com grande probabilidade de serem sarcomas, o papel da biópsia guiada por tomografia computadorizada é controverso, porém deve ser biopsiado em caso de diagnóstico incidental durante laparotomia exploradora.
No diagnóstico, a tomografia computadorizada é o exame de escolha, embora a ressonância nuclear magnética e exames intestinais contrastados possam ser úteis. A biópsia é útil para descartar linfoma ou metástase e para orientar a terapia cirúrgica, baseada na histologia do tumor. O estadiamento conclui-se com exames laboratoriais e radiografia de tórax. Cintilografia óssea deve ser indicada em casos de dor óssea ou alterações laboratoriais.
Apresentam-se como doença metastática no momento do diagnóstico em 20% dos casos. Avaliação anatômica e funcional renal para avaliar o rim contralateral é fundamental, já que o tratamento pode exigir a ressecção renal em 32-46% dos casos para se conseguir margens negativas (seguido de cólon em 25%, adrenal em 18%, pâncreas em 15% e baço em 10% dos casos). Atualmente, 20-40% dos tumores são irressecáveis por completo.
No estadiamento, todas os tumores de retroperitônio devem ser considerados lesões profundas, entretanto têm cursos clínicos variáveis, dependendo de seu tipo histológico e grau. A raridade de tais tumores, combinada com uma vasta gama de tipos histológicos, tem complicado seu conhecimento e afetado o desenvolvimento de novas terapias. Possuem uma grande tendência de recidiva local (mesmo no pós-operatório precoce) e disseminação para outros órgãos abdominais (como fígado), apresentando também recorrência à distância (pulmonar em até 20-30% dos casos), com relatos de recorrência em 5 anos de até 80%.
Como fatores prognósticos, as margens cirúrgica, seguida pelo grau do tumor, são as variáveis mais importantes na sobrevida livre de doença. A sobrevida depende diretamente da ressecção completa (margens negativas) e do grau. Sobrevida livre de doença em 5 e 10 anos de 63-65% e 43-56%, respectivamente, com risco de recidiva local de 25-50%.
| Lesões de Retroperitônio (adaptado de Lahey e Eckerson [1934], e Ackerman [1954]) |
||||
| Tumores | Tecido de Origem | Tumor benigno | Tumor maligno | |
| Adiposo | Lipoma | Lipossarcoma | ||
| Músculo liso | Leiomioma | Leiomiossarcoma | ||
| Músculo estriado | Rabdomioma | Rabdomiossarcoma | ||
| Conectivo | Fibroma | Fibrossarcoma | ||
| Vascular linfático | Hemangioma linfático | Linfangiossarcoma | ||
| Vascular sangüíneo | Hemangioma | Angiossarcoma | ||
| Hemangiopericitoma | ||||
| Mesenquima primitivo | Mixoma | Sarcoma de células claras (Mixossarcoma) | ||
| Histiócito | Histiocitoma | Histiocitoma Fibroso Maligno | ||
| Nervoso | Bainha nervosa | Neurofibroma não encapsulado | Schwannoma maligno | |
| SN Simpático | Ganglioneuroma | Ganglioneuroblastoma | ||
| Neuroblastoma | ||||
| Cromafim e adrenocortical | Paraganglioma | Paraganglioma maligno | ||
| Feocromocitoma | Feocromocitoma maligno | |||
| Incerto | Mesenquimoma | Mesenquimoma maligno | ||
| Xantogranuloma | Xantogranuloma maligno | |||
| Restos embrionários | Teratoma benigno | Teratoma maligno | ||
| Cordoma benigno | Cordoma maligno | |||
| Outras malignidades | Tumor de células germinativas, primário ou metastático | |||
| Tumores indiferenciados ou metastáticos | ||||
| Cistos | Wolffianos (origem urogenital) | |||
| Quilosos (origem linfática) | ||||
| Dermóides | ||||
| Mesocólicos | ||||
| Parasitários | ||||
| Traumáticos | ||||
| Massas não neoplásicas | Abscessos | |||
| Hematomas | ||||
| Fibrose retroperitoneal | ||||
| Tipos Histológicos de Sarcomas de Partes Moles (segundo TNM 2004) |
| Sarcoma alveolar de partes moles |
| Sarcoma epitelióide |
| Condrossarcoma extra-esquelético |
| Osteossarcoma extra-esquelético |
| Sarcoma de Ewing extra-esquelético |
| Tumor neuroectodérmico primitivo (PNET) |
| Fibrossarcoma |
| Leiomiossarcoma |
| Lipossarcoma |
| Histiocitoma fibroso maligno |
| Hemangiopericitoma maligno |
| Mesenquimoma maligno |
| Tumor maligno da bainha de nervo periférico |
| Rabdomiossarcoma |
| Sarcoma sinovial |
| Sarcoma SOE (sem outra especificação) |
Os seguintes tipos histológicos não são incluídos: sarcoma de Kaposi, dermatofibrossarcoma (protuberans), fibromatose (tumor desmóide) e sarcomas originados na duramater, cérebro, vísceras ocas ou órgãos parenquimatosos (com a exceção dos sarcomas de mama). O angiossarcoma, um sarcoma agressivo, é excluído porque sua história natural não é compatível com a classificação.
| Estadiamento TNM (2004) – Classificação Clínica | |||
| T | TX | O tumor primário não pode ser avaliado | |
| T0 | Não há evidência de tumor primário | ||
| T1 | Tumor com 5 cm ou menos em sua maior dimensão | ||
| T1a | Tumor superficial * | ||
| T1b | Tumor profundo* | ||
| T2 | Tumor com mais de 5 cm em sua maior dimensão | ||
| T2a | Tumor superficial * | ||
| T2b | Tumor profundo* | ||
| N | NX | Os linfonodos regionais não podem ser avaliados | |
| N0 | Ausência de metástase em linfonodos regionais | ||
| N1 | Metástase em linfonodos regionais | ||
| M | MX | A presença de metástase à distância não pode ser avaliada | |
| M0 | Ausência de metástase à distância | ||
| M1 | Metástase à distância | ||
* O tumor superficial é localizado exclusivamente acima da fáscia superficial, sem invasão desta; o tumor profundo é localizado ou exclusivamente sob a fáscia superficial ou superficialmente à fascia, com invasão ou penetração total desta. Os sarcomas retroperitoneal, mediastinal e pélvico são classificados como tumores profundos.
Deve haver confirmação histológica da doença e a divisão dos casos por tipo e grau histológico.
Categorias T (exame físico e diagnóstico por imagem), N (exame físico e diagnóstico por imagem) e M (exame físico e diagnóstico por imagem).
Os linfonodos regionais são aqueles referentes à localização do tumor primário. O envolvimento de linfonodos regionais é raro e os casos nos quais a condição nodal não pode ser avaliada, clinica ou patologicamente, devem ser considerados N0 ao invés de NX ou pNX.
Na Classificação Patológica (pTNM), as categorias pT, pN e pM correspondem às categorias T, N e M.
| Grupamento por Estádios | |
| Estádio | TNM |
| IA | T1a N0-X M0 Baixo grau T1b N0-X M0 Baixo grau |
| IB | T2a N0-X M0 Baixo grau T2b N0-X M0 Baixo grau |
| IIA | T1a N0-X M0 Alto grau T1b N0-X M0 Alto grau |
| IIB | T2a N0-X M0 Alto grau |
| III | T2b N0-X M0 Alto grau |
| IV | Qualquer T N1 M0 Qualquer grau Qualquer T Qualquer N M1 Qualquer grau |
| Graduação Histopatológica | ||
| Sistema de dois graus (TNM) | Sistema de 3 graus | Sistema de 4 graus |
| Baixo grau | Grau 1 | Grau 1 Grau 2 |
| Alto grau | Grau 2 Grau 3 |
Grau 3 Grau 4 |
Nota: Tumor neuroectodérmico primitivo (PNET) e sarcoma de Ewing extra-esquelético são classificados como de alto grau.
Tratamento
Se possível, o paciente deve ser visto por uma equipe mutidisciplinar. O objetivo desta estratégia é evitar ressecções maiores inapropriadas de outros tumores, como linfoma e tumores de células germinativas.
O tratamento é eminentemente cirúrgico, com ressecções extensas e com margens cirúrgicas amplas, mesmo que para isso seja necessária a ressecção de órgãos adjacentes, podendo ser seguido de radioterapia intraoperatória ou pós-operatória em casos de margens cirúrgicas insuficientes. Em alguns casos, indica-se radioterapia ou quimioterapia primárias, prévias à cirurgia. Em casos de recidivas operáveis, a terapia cirúrgica é indicada. Já nos casos de doenças irressecáveis ou tumores avançados, tratamentos paliativos como quimioterapia e/ou radioterapia estão indicados.
Seguimento
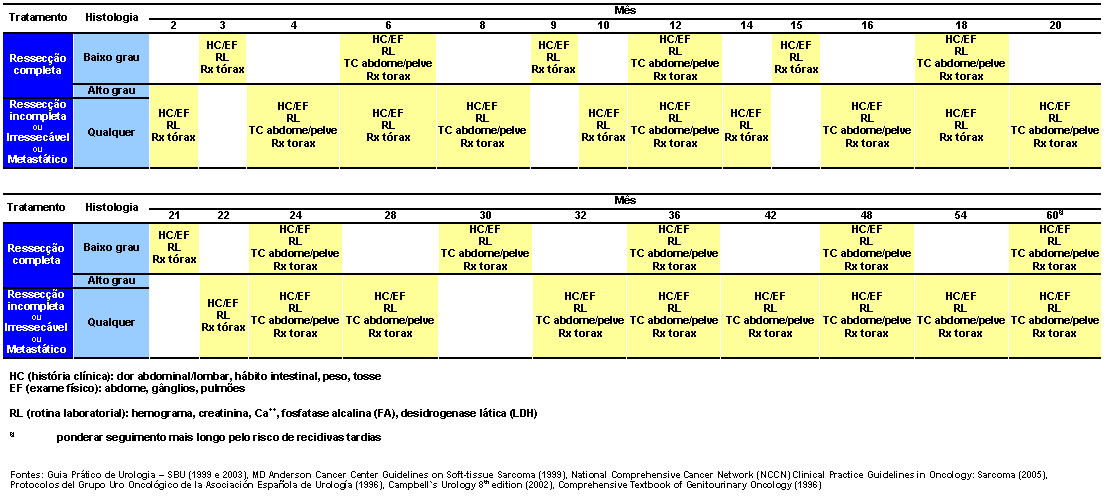
Fontes:
Campbell-Walsh Urology, 10th edition (2011)
Comprehensive Textbook of Genitourinary Oncology, 4th edition (2011)
National Comprehensive Cancer Network (NCCN) Clinical Practice Guidelines in Oncology (NCCN Guidelines®) – Soft Tissue Sarcoma – Version 2.2014
Links úteis para pacientes:
Soft Tissue Sarcoma